Chromatin state maps
2012-2015
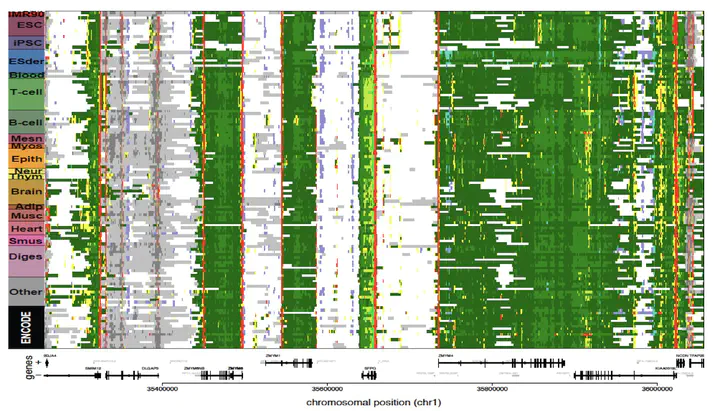
The reference human genome sequence set the stage for studies of genetic variation and its association with human disease, but up until 2015 epigenomic studies lacked a similar reference. To address this need, we generated the largest collection of human epigenomes for primary cells and tissues to date.
We performed an integrative analysis of 111 reference human epigenomes generated as part of the programme, profiled for histone modification patterns, DNA accessibility, DNA methylation and RNA expression. Focusing on histone tail modifications, we defined extensive chromatin state models and maps. By integrating these maps with chromatin accessibility data obtained from DNase-seq experiments, we established global maps of regulatory elements.
We showed that disease- and trait-associated genetic variation is preferentially localized in tissue-specific regulatory elements, revealing biologically relevant cell types for diverse human traits, and providing a resource for interpreting the molecular basis of human disease.
Taken together, our results demonstrated the central role of epigenomic information for understanding gene regulation, cellular differentiation and human disease.
Kundaje, Meuleman, Ernst, Bilenky et al., Nature (2015)
Work with Anshul Kundaje, Manolis Kellis, and others.
Wouter Meuleman
Principal Investigator
Altius Institute
Affiliate Associate Professor
University of Washington
My research interests include computational (epi)genomics, genome organization, and data visualization