Data saliency - epilogos
2012-2019
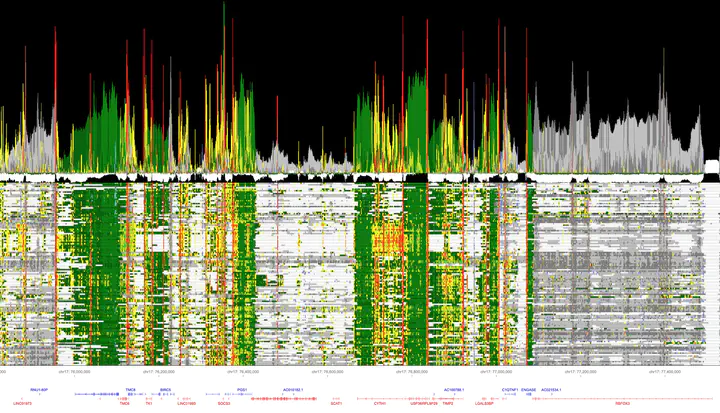
DNA is organized in the cell nucleus by way of a chromatin structure, which is shaped and maintained by many chromatin marks, such as histone tail modifications (e.g., H3K4me1), transcription factors (e.g., GATA1) and chromatin modifiers (e.g., EZH2). The genome-wide occurrence of such chromatin marks can be captured using assays such as Chromatin Immunoprecipitation Sequencing (ChIP-Seq). Chromatin states describe the combinatorial occurrence of multiple chromatin marks, defined using software such as ChromHMM and Segway.
Chromatin states across 100s of biosamples can capture the dynamics of chromatin marks. Although this provides clues to the function of genomic regions, it results in large datasets that remain hard to navigate.
By modeling the information content, we transform multi-biosample chromatin state data into intuitive readouts called epilogos. This process is analogous to how motif logos are derived from DNA or protein sequence alignments, and opens up new possibilities for studying chromatin mark dynamics in large genome-wide datasets.
Wouter Meuleman
Principal Investigator
Altius Institute
Affiliate Associate Professor
University of Washington
My research interests include computational (epi)genomics, genome organization, and data visualization